Python Names functions & Modules¶
- A Python name consists of an arbitrary number of letters, underscores, and digits.
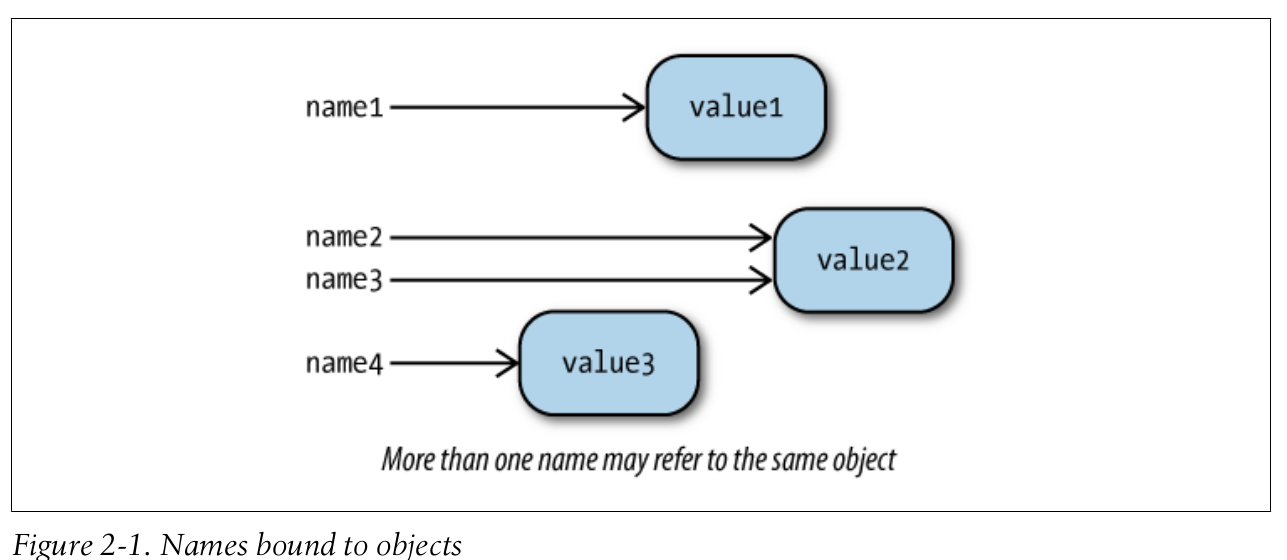
- A Python program consists of a series of statements.
- An assignment statement binds a name to an object.
name = value
In [1]:
aaseq1 = 'MNKMDLVADVAEKTDLSKAKATEVIDAVFA'
In [2]:
aaseq2 = 'AARHQGRGAPCGESFWHWALGADGGHGHAQPPFRSSRLIGAERQPTSDCRQSLQ'
In [3]:
aaseq1
Out[3]:
In [5]:
len(aaseq2)
Out[5]:
In [6]:
aaseq3
In [8]:
aaseq3 = aaseq1
In [9]:
aaseq3
Out[9]:
- Assignment statements may be chained together. This feature is used primarily to bind several names to some initial value:
a = b = c = 0 - Another variation on assignment statements is a bit of shorthand for arithmetic updates.
a = a + 1
a += 1 - This is called an augmented assignment statement.
- += , -= , *= , /= , //= , %= , and **= .
Defining Function¶
- New functions are defined with function definition statements.
- A definition is a compound statement, meaning it comprises more than one line of code.
- The first line of each compound statement ends with a colon.
- Subsequent lines are indented relative to the first. The standard practice is to indent by four spaces
In [11]:
def name(parametr_list):
body
- Some functions are intended to return a value and some aren’t.
- A
returnstatement is used to return a value from a function. - The word
returnis usually followed by an expression whose value is returned as the value of the function. - Occasionally a return is not followed by an expression, in which case the value returned is
None. - All functions return a value, whether or not they contain a return statement: if the function finishes executing without encountering a return , it returns None .
- A function’s body must contain at least one statement.
- The do-nothing statement that is equivalent to the no-value
Noneis the simple statementpass.
In [1]:
def fn():
pass
fn()
In [13]:
# A simple function for recognizing a binding site
def recognition_site(base_seq, recognition_seq):
return base_seq.find(recognition_seq)
In [15]:
recognition_site("ATGCATGCATGC","GCAT")
Out[15]:
Compare the length of the sequence string to the sum of the number of Ts, Cs, As, and Gs in the sequence string. If the length is equal to that sum, the function returns True ; otherwise, there is a character in the parameter sequence that isn’t a valid base character, and the result will be False .
In [2]:
def validate_base_sequence(base_sequence):
"""Return True if string base_seqeunce contains only
upper-or lovercase T,C,A and G characters, otherwise False"""
seq = base_sequence.upper()
return len(seq) == (seq.count('T') + seq.count('C') +
seq.count('A') + seq.count('G'))
In [3]:
help(validate_base_sequence)
In [4]:
validate_base_sequence('tattattat')
Out[4]:
In [5]:
validate_base_sequence('atgcwrqatgc')
Out[5]:
Compute the GC content of a given DNA sequence represented as a string.
In [20]:
def gc_content(base_seq):
"""Returns the percentage of G and C chracters in base_seq"""
seq = base_seq.upper()
return (seq.count('G') + seq.count('C'))/ len(seq)
In [5]:
seq50 = 'AACCTTGG'
In [6]:
seq75 = 'ATCCCGGG'
In [7]:
seq40 = 'ATATTTCGCG'
In [8]:
gc_content(seq50)
Out[8]:
In [9]:
gc_content(seq75)
Out[9]:
In [10]:
gc_content(seq40)
Out[10]:
Assertions¶
- An assertion statement tests whether an expression is true or false, causing an error if it is false.
assert expression - A two-expression assertion statement takes two arguments: an expression to evaluate and an expression to use in the error report if the first expression is false.
assert expression1, expression2
In [1]:
assert 1 == 2
In [2]:
assert 1 == 2, 'invalid arguments'
In [4]:
def gc_content(base_seq):
"""Returns the precentage of G and C characters in base_seq"""
assert validate_base_sequence(base_seq), \
'argumnet has invalid arguments'
seq = base_seq.upper()
return ((base_seq.count('G') + base_seq.count('C'))/
len(base_seq))
Default Parameter Values¶
- Python provides a way to assign a default value to the parameter that will be used if no explicit value is included in a call to the function.
- Some parameters of some functions are optional.
- A function definition designates an optional parameter by assigning it a default value in the parameter list.
- Make
validate_base_sequencemore flexible by giving it the ability to handle RNA sequences too.
In [11]:
def validate_base_sequence(base_sequence, RNAflag):
"""Returns True if the string base_sequence contains only
upper or lowercase T (or U, if RNAflag), C, A, G characters
otherwise False"""
seq = base_sequence.upper()
return len(seq) == (seq.count('U' if RNAflag else 'T') +
seq.count('C') + seq.count('A') +
seq.count('G'))
In [12]:
validate_base_sequence('ATGC', False)
Out[12]:
In [13]:
validate_base_sequence('ATGC', True)
Out[13]:
In [14]:
validate_base_sequence('AUCG', True)
Out[14]:
In [15]:
def validate_base_sequence(base_sequence, RNAflag=False):
"""Returns True if the string base_sequence contains only
upper or lowercase T (or U, if RNAflag), C, A, G characters
otherwise False"""
seq = base_sequence.upper()
return len(seq) == (seq.count('U' if RNAflag else 'T') +
seq.count('C') + seq.count('A') +
seq.count('G'))
In [16]:
validate_base_sequence('ATCG')
Out[16]:
In [17]:
validate_base_sequence('AUGC', True)
Out[17]:
Using Modules¶
- Apart from the primitives, Python offers a large selection of optional types, functions and methods, they are defined by module files placed in a library directory of Python installation.
- Modules can be installed from external sources.
- Module file contains Python statements along with documentation describing its contents and purpose.
Importing¶
- The basic form of the import statement loads a module into the Python environment and makes its name available in the namespace into which it was imported
import name
name is name of module - no path or extention. - Module contents are accessed using the dot notation used form method calls.
- Modules may have submodules, referenced by dot notation.
os.path
In [1]:
import os # provides an interface to the computer’s operating system.
In [2]:
os
Out[2]:
In [3]:
os.getcwd()
Out[3]:
Selective import¶
- Another form of
importallows import of specific names from the module
from modulename import name1, name2, ...
from modulename import actualname as yourname
from modulename import *
In [5]:
from sys import version
In [6]:
version
Out[6]:
random module¶
- One useful module is random , which provides various ways to generate random numbers.
- The function
random.randinttakes two integer arguments and returns a random integer in the range of the first to the second inclusive.
Generate a random codon¶
In [6]:
from random import randint
In [7]:
def random_base(RNAflag = False):
return ('UCAG' if RNAflag else 'TCAG')[randint(0,3)]
In [8]:
def random_codon(RNAflag = False):
return random_base(RNAflag) + random_base(RNAflag) + \
random_base(RNAflag)
In [17]:
random_codon()
Out[17]:
Simulating a single-base mutation¶
In [20]:
def replace_base_randomly(base_seq):
"""Return a sequence with the base at a randomly selected position
of base_seq replaced by a base chosen randomly from the three bases
that are not at that position"""
print(base_seq)
position = randint(0, len(base_seq) - 1)
print(position)
bases = 'TCAG'.replace(base_seq[position],'')
print(bases)
return (base_seq[0:position])+ bases[randint(0,2)] \
+ base_seq[position+1:]
In [21]:
replace_base_randomly('ATGCATGCATGC')
Out[21]: